2021年7月,我校第一附属医院季晶教授团队在《Nature Communications》上发表了题为“Prokineticin-2 prevents neuronal cell deaths in a model of traumatic brain injury”的研究论文,揭示了 Prok2 在神经元铁死亡中的调节作用和机制。
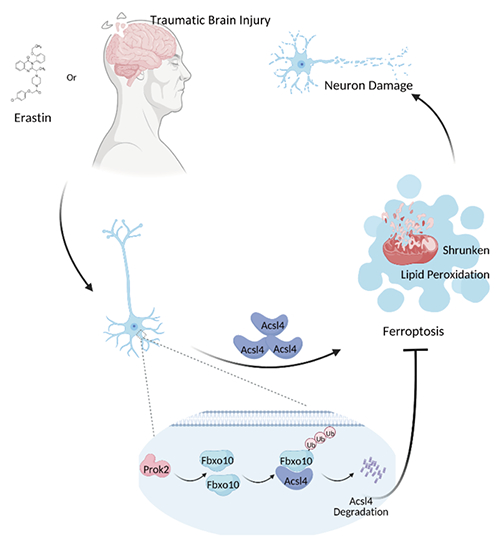 |
图1 Prok2相关信号通路阻止颅脑外伤后神经元铁死亡的机制模式图

在人类和小鼠受控皮质冲击 (CCI) 模型中,以及暴露于机械拉伸和亲铁性神经毒素的初级皮质神经元中,在创伤性脑损伤 (TBI)后Prok2表达被迅速诱导。
Prok2 增加 Fbxo10(一种与 Acsl4 结合的泛素连接酶)的水平,从而促进 Acsl4 泛素化和降解。
Prok2 驱动的级联反应可减轻铁死亡,保留线粒体功能,并保护神经元免受 TBI。
创伤性脑损伤 (TBI) 是全球死亡和残疾的主要原因。TBI发展为两个阶段的过程。第一阶段是脑组织的机械损伤,直接导致神经元死亡。第二阶段包括由多种机制引起的周围微环境的损害和恶化的扩散——促炎反应、氧化应激、局部缺氧/复氧和神经毒性物质的积累,由于细胞凋亡或坏死性炎症程序之一(如坏死性凋亡、细胞焦亡和自噬)的时间依赖性激活,最终导致神经元死亡。
铁死亡是最近发现的一种细胞死亡程序,由磷脂氢过氧化物(尤其是 HOO-花生四烯酸-PE)的积累定义,由铁依赖机制和硫醇调节不足催化。酰基辅酶 A 合成酶长链家族成员 4 (Acsl4) 是花生四烯酸 (AA)-PE 生物合成中的关键酶,已被证明有助于铁死亡。Prokineticins 是新发现的趋化因子,在免疫系统和炎症性疾病中具有关键作用 ,并且可能参与神经系统疾病的发病机制。Prokineticin-2 (Prok2) 是 prokineticin 家族的成员,目前尚未研究 Prok2 在 TBI 中的功能,特别是在神经元细胞死亡的背景下。
研究人员首先发现,TBI患者脑组织中Prok2 水平显着增加,此外,对原代皮层神经元进行了体外机械拉伸以及用特定的铁死亡诱导剂 Erastin处理细胞后也会增加 Prok2 的表达。表明Prok2 在脑损伤后上调并参与了铁死亡的调节。进一步探讨了Prok2 作为原代神经元细胞铁死亡调节剂的作用发现,Prok2的过度表达可降低 Erastin 或拉伸诱导的神经元细胞毒性和脂质过氧化,在 Erastin 或拉伸治疗后,含氧 AA 代谢物、15-羟基二十碳四烯酸 (15-HETE) 和 12-羟基二十碳四烯酸 (12-HETE) 的水平显着增加,而Prok2的上调降低了这些代谢物的水平。此外,Prok2 上调可以保护Erastin或拉伸处理后的线粒体功能,且这种保护功能是Acsl4依赖性介导的,在过度表达 Prok2 的铁死亡细胞中 Acsl4 表达降低。
Prok2 介导的 Acsl4 含量降低是抑制神经元铁死亡的重要因素,研究人员接着探索了 Prok2 在体外下调 Acsl4 的可能机制,发现Prok2 过表达增强了 Acsl4 泛素化并可能引导 Acsl4 朝向其蛋白酶体降解,而这一过程是通过泛素连接酶Fbxo10驱动的。Prok2 过表达通过促进 Fbxo10 的表达和加速 Acsl4 泛素化/降解来减轻铁死亡。
最后,研究人员在体内验证了所提出机制的有效性,在受控皮质撞击(CCI)损伤之前注射腺相关病毒 Prok2 的小鼠通过调节Acsl4表现出神经元变性减少并改善运动和认知功能,这可以通过敲低 Fbxo10 得到抑制。
综上所述,该研究确定了Prok2在TBI后病理生理学中的神经保护作用,这种保护策略可以通过下调Acsl4来增强。具体来说, Prok2的过度表达通过增加Fbxo10的表达和促进Acsl4泛素化/降解来减轻由Erastin诱导的细胞损伤。
